Feature Selection and Feature Engineering with multiDEGGs
Elisabetta Sciacca, Myles Lewis
Source:vignettes/Feature_Selection.Rmd
Feature_Selection.RmdFeature Selection and Feature Engineering with multiDEGGs in Nested Cross-Validation
In computational biology applications involving high-throughput data, researchers commonly encounter situations where the number of potential predictors far exceeds the available sample size. This dimensional challenge requires careful feature selection strategies for both mathematical and clinical reasons.
Standard feature selection methods typically evaluate predictors individually, identifying those variables that show the strongest univariate associations with the outcome variable (such as through t-tests or Wilcoxon tests). While effective, this approach overlooks the interconnected nature of biological systems, where
Feature engineering represents a complementary strategy that creates new predictors by combining or transforming existing variables. In biology, such approach can be used to capture higher-order information that reflects the interconnected nature of molecular processes. For instance, the ratio between two genes may provide more discriminative power than either gene expression level independently, particularly when their relative balance is disrupted in disease states.
The informative content encoded in differential interactions, combined with multiDEGGs’ ability to identify only literature-validated differential relationships, makes it particularly well-suited for both individual feature selection and guided creation of engineered predictors in machine learning. Such approach has potential to overcome the limitations of conventional algorithms which may select individual predictors without clear biological significance, compromising both the interpretability and clinical credibility of the resulting models.
Why Nested Cross-Validation for Feature Engineering?
It is crucial that feature selection and modification is conducted
exclusively on training data within cross-validation loops to prevent
information leakage from the test set. The nestedcv package
enables the nested modification of predictors within each outer fold,
ensuring that the attributes learned from the training part are applied
to the test data without prior knowledge of the test data itself.
The selected and combined features, and corresponding model, can then be
evaluated on the hold-out test data without introducing bias.
Both nestcv.glmnet and nestcv.train from
nestedcv accept any user-defined function that filters or
transforms the feature matrix by passing the function name to the
modifyX parameter.
The multiDEGGs package provides two specialized functions for
this purpose.
multiDEGGs_filter(): Pure Differential Network-Based Selection
The multiDEGGs_filter() function performs feature
selection based entirely on differential network analysis. It identifies
significant differential molecular interactions and can return either
the interaction pairs alone or both pairs and individual variables
involved in those interactions.
Key Parameters
When using multiDEGGs_filter(), you can control the
following parameters through modifyX_options:
-
keep_single_genes(logical, defaultFALSE): Controls whether to include individual genes from significant pairs in addition to the pairs themselves -
nfilter(integer, defaultNULL): Maximum number of predictors to return. WhenNULL, all significant interactions found are included
Usage Examples
Basic Usage: Pairs Only
library(multiDEGGs)
library(nestedcv)
data("synthetic_metadata")
data("synthetic_rnaseqData")
# Regularized linear model with interaction pairs only
fit.glmnet <- nestcv.glmnet(
y = as.numeric(synthetic_metadata$response),
x = t(synthetic_rnaseqData),
modifyX = "multiDEGGs_filter",
modifyX_options = list(
keep_single_genes = FALSE,
nfilter = 20
),
modifyX_useY = TRUE,
n_outer_folds = 5,
n_inner_folds = 6,
verbose = FALSE
)
summary(fit.glmnet)#> Nested cross-validation with glmnet
#> No filter
#> Modifier: multiDEGGs_filter
#> Outer loop: 5-fold CV
#> Inner loop: 6-fold CV
#> 100 observations, 14 predictors
#>
#>
#> Final parameters:
#> lambda alpha
#> 0.05894 0.10000
#>
#> Final coefficients:
#> (Intercept) TNF:TNFRSF1A AKT2:MTOR IL1B:IL1R2 FASLG:FAS TGFB3:TGFBR1
#> 1.823874 -0.193020 -0.119887 0.052089 -0.035947 -0.033527
#> MAP2K2:MAPK3 FANCD2:FAN1
#> -0.021308 -0.008862
#>
#> Result:
#> RMSE R.squared Pearson.r^2 MAE
#> 0.47302 0.08148 0.09173 0.44153Including Individual Genes (keep_single_genes = TRUE)
# Random forest model including both pairs and individual genes
fit.rf <- nestcv.train(
y = synthetic_metadata$response,
x = t(synthetic_rnaseqData),
method = "rf",
modifyX = "multiDEGGs_filter",
modifyX_options = list(
keep_single_genes = TRUE,
nfilter = 30
),
modifyX_useY = TRUE,
n_outer_folds = 5,
n_inner_folds = 6,
verbose = FALSE
)
fit.rf$summary
# Plot ROC on outer folds
plot(fit.rf$roc)#> Reference
#> Predicted Non_responder Responder
#> Non_responder 57 2
#> Responder 1 40
#>
#> AUC Accuracy Balanced accuracy
#> 0.9979 0.9700 0.9676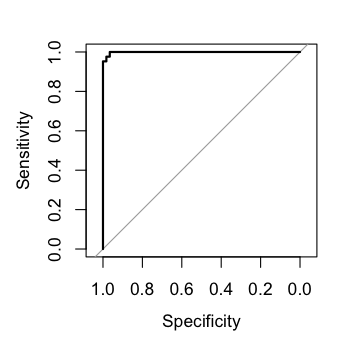
How nfilter works with keep_single_genes
- When
keep_single_genes = FALSE:nfilterlimits only the number of interaction pairs returned - When
keep_single_genes = TRUE:nfilterlimits the combined count of unique individual genes plus interaction pairs. The function prioritizes pairs by significance and adds individual genes as needed until the limit is reached
multiDEGGs_combined_filter(): Hybrid Statistical and Network-Based Selection
The multiDEGGs_combined_filter() function combines
traditional statistical feature selection with differential network
analysis. This hybrid approach allows you to benefit from both
conventional univariate selection methods and the biological insights
from interaction analysis.
Key Parameters
-
filter_method(character): Statistical method for single feature selection.
Options:"ttest","wilcoxon","ranger","glmnet","pls" -
nfilter(integer): Maximum number of features to select -
dynamic_nfilter(logical): Controls hownfilteris applied (see detailed explanation below) -
keep_single_genes(logical): Whendynamic_nfilter = TRUE, determines whether to include individual genes from multiDEGGs pairs
Dynamic vs. Balanced Selection Modes
Dynamic Selection (dynamic_nfilter = TRUE)
In dynamic mode, the function: 1. Selects nfilter single
genes using the chosen statistical method 2. Adds ALL significant
interaction pairs found by multiDEGGs 3. Total predictors =
nfilter single genes + number of significant pairs
This mode allows the feature space to expand based on the biological complexity discovered in each fold.
# Dynamic selection with t-test for single genes
fit.dynamic <- nestcv.glmnet(
y = as.numeric(synthetic_metadata$response),
x = t(synthetic_rnaseqData),
modifyX = "multiDEGGs_combined_filter",
modifyX_options = list(
filter_method = "ttest",
nfilter = 20,
dynamic_nfilter = TRUE,
keep_single_genes = FALSE
),
modifyX_useY = TRUE,
n_outer_folds = 5,
n_inner_folds = 6,
verbose = FALSE
)Balanced Selection (dynamic_nfilter = FALSE)
In balanced mode, the function:
1. Allocates approximately half of nfilter to interaction
pairs
2. Fills remaining slots with single genes from the statistical
filter
3. Maintains consistent total number of predictors across all folds
This mode ensures a fixed feature space size while balancing single genes and interactions.
# Balanced selection with Wilcoxon-test importance
fit.balanced <- nestcv.train(
y = synthetic_metadata$response,
x = t(synthetic_rnaseqData),
method = "rf",
modifyX = "multiDEGGs_combined_filter",
modifyX_options = list(
filter_method = "wilcoxon",
nfilter = 40,
dynamic_nfilter = FALSE
),
modifyX_useY = TRUE,
n_outer_folds = 5,
n_inner_folds = 6,
verbose = FALSE
)Available Statistical Methods
-
"ttest": Two-sample t-test for differential expression -
"wilcoxon": Wilcoxon rank-sum test (non-parametric alternative to t-test) -
"ranger": Random Forest variable importance scoring (therangerpackage must be installed first) -
"glmnet": Elastic net regularization coefficients -
"pls": Partial Least Squares variable importance
Practical considerations
Before implementing multiDEGGs in your machine learning pipeline, it’s highly recommended to first run a preliminary analysis on your complete dataset to assess the number of differential interactions detected. This exploratory step can guide your choice of approach and parameter settings.
If multiDEGGs identifies only a small number of differential interactions (e.g., fewer than 10-20 pairs), these features alone may lack sufficient predictive power. In such cases, consider:
- Using
multiDEGGs_combined_filter()to integrate network-based features with traditional statistical selection methods - Setting
keep_single_genes = TRUEinmultiDEGGs_filter()to include individual genes involved in the differential pairs - Adjusting the
percentile_vectoror significance thresholds in the initial multiDEGGs analysis to potentially capture more interactions
Conversely, if a large number of differential interactions are
detected, multiDEGGs_filter() alone may provide sufficient
feature diversity for effective model training.
Feature Engineering Details
Both functions create ratio-based features from significant gene
pairs (Gene A / Gene B), which capture the relative expression
relationships that drive differential network connectivity. The
predict methods automatically handle the feature
transformation for both training and test data within each
cross-validation fold, ensuring no information leakage.
Note: If no significant differential interactions are found in a particular fold, both functions automatically fall back to t-test-based selection to ensure robust performance across all scenarios. This fallback is indicated by a printed “0” during execution.
Citation
citation("multiDEGGs")
#> To cite package 'multiDEGGs' in publications use:
#>
#> Sciacca E, Wang S, Pitzalis C, Lewis M (2026). "multiDEGGs: Single or
#> Multiomic Differential Network Analysis for Biomarker Discovery and
#> Feature Engineering for Predictive Modeling." _Computational and
#> Structural Biotechnology Journal_, *35*(1), 0001.
#> doi:10.34133/csbj.0001 <https://doi.org/10.34133/csbj.0001>.
#>
#> A BibTeX entry for LaTeX users is
#>
#> @Article{,
#> title = {multiDEGGs: Single or Multiomic Differential Network Analysis for Biomarker Discovery and Feature Engineering for Predictive Modeling},
#> author = {Elisabetta Sciacca and Susan S. Wang and Costantino Pitzalis and Myles J. Lewis},
#> journal = {Computational and Structural Biotechnology Journal},
#> year = {2026},
#> volume = {35},
#> number = {1},
#> pages = {0001},
#> doi = {10.34133/csbj.0001},
#> }